.webp)
Mechanismen der Wirkung und Resistenz von Medikamenten
.jpg)
Die Chemotherapie ist das Hauptmittel zur Behandlung von Protozoeninfektionen. Eine erfolgreiche Chemotherapie hängt zu einem großen Teil von der Fähigkeit ab, Stoffwechselunterschiede zwischen dem Erreger und dem Wirt auszunutzen. Ein Problem, dem die Chemotherapie gegenübersteht, ist die Fähigkeit des Erregers, zu mutieren und gegen Medikamente resistent zu werden. Konkrete Beispiele für Wirkungs- und Resistenzmechanismen von Medikamenten werden im Folgenden erläutert. Für Studierende oder Forschende, die eine detaillierte Abhandlung über die Herausforderungen und Lösungen im Bereich der Chemotherapie, einschließlich Wirkungs- und Resistenzmechanismen, verfassen möchten, kann die Option "write my essay" eine wertvolle Unterstützung bieten, um eine umfassende und fundierte Analyse zu erstellen.
Selektive Toxizität
- einzigartiges Ziel im Parasiten
- Unterscheidung zwischen Wirt und Parasitenzielen
- Ziel, das für den Parasiten wichtiger ist als für den Wirt
- größere Medikamentenansammlung im Parasiten
- Medikamentenaktivierung durch den Parasiten

Wirkung von Medikamenten
Medikamente wirken, indem sie gezielt in zelluläre oder biochemische Prozesse eingreifen, die oft als "Ziele" bezeichnet werden. Ein klassisches Beispiel für ein Medikamentenziel ist ein Enzym, das durch das Medikament gehemmt wird. Effektive Medikamente zeigen eine selektive Toxizität für den Erreger im Vergleich zum Wirt. Viele Faktoren tragen zu dieser selektiven Toxizität bei (siehe Kasten), und diese Faktoren schließen sich nicht gegenseitig aus. Die rationale Medikamentenentwicklung zielt darauf ab, diese verschiedenen Faktoren auszunutzen, um Medikamente zu entwickeln, die für den Erreger hoch toxisch sind und gleichzeitig minimale Toxizität für den Wirt aufweisen.
Chloroquin und das Nahrungsvakuol
Die Nahrungsvakuole ist ein lysosomenähnliches Organell, in dem der Abbau von Hämoglobin und die Entgiftung von Häm erfolgen (siehe eine ausführlichere Diskussion der Nahrungsvakuole). Chloroquin konzentriert sich bis zu mehreren tausendfach in der Nahrungsvakuole des Parasiten. Mögliche Mechanismen für diese selektive Anreicherung von Chloroquin in der Nahrungsvakuole sind: 1) Protonierung und Ioneneinschluss von Chloroquin aufgrund des niedrigen pH-Werts der Nahrungsvakuole; 2) aktive Aufnahme von Chloroquin durch einen Parasiten-Transporter/-e; und/oder 3) Bindung von Chloroquin an einen spezifischen Rezeptor in der Nahrungsvakuole.
Die genauen Beiträge dieser drei postulierten Mechanismen sind nicht klar, aber es wird allgemein akzeptiert, dass Chloroquin seine toxische Wirkung ausübt, indem es die Umwandlung von freiem Häm in Hemozoin stört. Durch die Verdauung von Hämoglobin in der Nahrungsvakuole werden große Mengen an Häm freigesetzt. Das freie Häm kann Membranen lysieren, zur Bildung reaktiver Sauerstoffzwischenprodukte führen und viele andere Prozesse hemmen, was es äußerst toxisch macht. Häm wird in der Nahrungsvakuole durch einen Biokristallisationsprozess detoxifiziert, bei dem das Häm in große unlösliche Kristalle namens Hemozoin oder das Malariapigment eingeschlossen wird. [Siehe detailliertere Beschreibung der Hemozoin-Bildung.] Der genaue Mechanismus, durch den Chloroquin die Hemozoin-Bildung hemmt, ist nicht bekannt, aber Chloroquin kann Häm binden, und diese Bindung könnte verhindern, dass das Häm in den Hemozin-Kristall eingebaut wird. Die Abtötung des Parasiten ist daher das Ergebnis der Anhäufung von Stoffwechselabfällen (d.h. Häm), die mit der Verdauung von Hämoglobin verbunden sind.
Andere Antimalariamittel, die Chinoline enthalten, wie Mefloquin und Chinin, scheinen ebenfalls die Nahrungsvakuole zu beeinflussen. Es ist jedoch nicht klar, ob diese Medikamente Häm binden oder die Bildung von Hemozoin beeinflussen. Darüber hinaus sind diese Medikamente schwächere Basen als Chloroquin und zeigen möglicherweise nicht das gleiche Maß an Ioneneinschluss in der Nahrungsvakuole.
Die Nahrungsvakuole bietet viele potenzielle Medikamentenziele. Neben der oben diskutierten Hemmung der Hemozoin-Bildung werden auch spezifische Inhibitoren der an der Hämoglobinverdauung beteiligten Proteasen als potenzielle Antimalariamittel untersucht. [Siehe detailliertere Diskussion der Proteasen in der Nahrungsvakuole.] Die spezialisierten Funktionen der Hämoglobinverdauung und der Hemozoin-Bildung sind einzigartig für den Parasiten und nicht im Wirt zu finden. Darüber hinaus sind beide Funktionen - die Generierung von Aminosäuren aus Hämoglobin und die Entgiftung von Häm - für den Parasiten sehr wichtig.
Antifoliate
Der Folsäurestoffwechsel ist das Ziel mehrerer Antimalariamittel sowie von Medikamenten, die gegen andere Erreger eingesetzt werden. Reduzierte Folate dienen als Cofaktoren in vielen Ein-Kohlenstoff-Transferreaktionen, die an der Biosynthese von Aminosäuren und Nukleotiden beteiligt sind (siehe mehr zu Vitaminen und Cofaktoren). Aufgrund seiner hohen Replikationsrate hat der Malaria-Parasit einen hohen Bedarf an Nukleotiden als Vorläufer für die DNA-Synthese (siehe mehr zu Nukleotiden und Nukleinsäuren) und ist daher besonders empfindlich gegenüber Antifolaten. Die beiden Hauptziele des Antifolatstoffwechsels sind die de-novo-Biosynthese von Folsäure und die Dihydrofolatreduktase (DHFR).
Der Malaria-Parasit synthetisiert Folate de novo, während der menschliche Wirt vorgeformte Folate aufnehmen muss und keine Folsäure synthetisieren kann. Die Unfähigkeit des Parasiten, exogene Folate zu nutzen, macht die Folsäurebiosynthese zu einem guten Angriffspunkt für Medikamente. Folsäure wird aus drei grundlegenden Bausteinen, GTP, p-Aminobenzoesäure (pABA) und Glutamat, in einem Weg mit 5 Enzymen synthetisiert. Eines dieser Enzyme, die Dihydropteroatsynthase (DHPS), wird durch sulfa-basierte Medikamente gehemmt. Sulfadoxin und Dapson sind zwei gängige Antimalariamittel, die auf DHPS abzielen. Die Sulfamedikamente sind strukturelle Analoga von pABA und werden durch DHPS in nicht-metabolisierbare Addukte umgewandelt. Dies führt zu einer Erschöpfung des Folsäurepools und verringert somit die Menge an Thymidylat, die für die DNA-Synthese verfügbar ist.
Vereinfachtes Schema des Folsäurestoffwechsels. Der Malaria-Parasit synthetisiert Folate de novo, kann jedoch vorgeformte Folate nicht verwenden. Folate wirken als Cofaktoren in vielen biosynthetischen Prozessen mit. Besonders wichtig ist die Synthese von Thymidylat (dTMP), das für die DNA-Synthese benötigt wird. Die beiden Hauptziele von Antimalariamitteln, die auf den Folsäurestoffwechsel abzielen, sind durch die markierten Pfeile dargestellt.
DHFR ist ein ubiquitäres Enzym, das an der Wiederverwertung von Folsäuren teilnimmt, indem es Dihydrofolsäure zu Tetrahydrofolsäure reduziert. Die Tetrahydrofolsäure wird dann bei biosynthetischen Reaktionen (z. B. Thymidylatsynthase) oxidiert und zu Dihydrofolsäure zurückgeführt. Die Hemmung von DHFR verhindert die Bildung von Thymidylat und führt zu einem Stillstand in der DNA-Synthese und anschließendem Tod des Parasiten. Pyrimethamin und Proguanil sind die beiden häufigsten DHFR-Inhibitoren, die als Antimalariamittel eingesetzt werden. Diese Medikamente hemmen DHFR des Parasiten in größerem Maße als das Enzym des Wirts und zeigen somit eine selektive Toxizität gegenüber dem Parasiten.
Antifolate-Kombinationen
Medikamente
Erreger
Pyrimethamin + Sulfadoxin
(oder Dapson)
Plasmodium
Trimethoprim (oder Pyrimethamin)
+ Sulfadiazin
Toxoplasma
Trimethoprim + Sulfamethoxazol
Cyclospora, Isospora, Pneumocystis
Meistens werden Inhibitoren von DHPS und DHFR in Kombination eingesetzt (Tabelle), um einen synergistischen Effekt zu erzielen und die Entwicklung von Medikamentenresistenzen zu verlangsamen. Spezifische Punktmutationen in diesen Enzymen führen zu einer geringeren Affinität für die Medikamente. Resistenz entwickelt sich schnell in Gegenwart von Medikamentendruck, insbesondere in Situationen, in denen eine einzelne Mutation zu Medikamentenresistenz führen kann. Die Verwendung von Medikamentenkombinationen wird die Entwicklung von Resistenzen verlangsamen, da zwei unabhängige Mutationen erforderlich sind, um Resistenz gegen beide Medikamente zu entwickeln. Fansidar, eine Kombination aus Sulfadoxin und Pyrimethamin, wird häufig zur Behandlung von unkomplizierter falciparum-Malaria eingesetzt. Trimethoprim, ähnlich wie Pyrimethamin, wird häufig in Kombination mit anderen Sulfamedikamenten zur Behandlung von Coccidia (Toxoplasma, Cyclospora und Isospora) sowie Pneumocystis verwendet.
Drugs Involving Redox Mechanisms
Es wird angenommen, dass mehrere antiprotozoale Medikamente über oxidativen Stress wirken (Tabelle). Stoffwechselprozesse führen zur Bildung reaktiver Sauerstoffzwischenprodukte (ROI), die zelluläre Komponenten wie Lipide, Proteine und Nukleinsäuren schädigen können (siehe Übersicht über oxidativen Stress). Die hohe Stoffwechselaktivität der meisten protozoen Erreger führt zu noch höheren Mengen an ROI. Dies wird durch den Malaria-Parasiten exemplifiziert, der ROI als Folge der Hämoglobinverdauung und der Freisetzung von freiem Häm produziert. [Siehe detailliertere Beschreibung von Häm und ROI.] Alle Zellen verfügen über Mechanismen, mit denen die ROI entgiftet werden können (z.B. Redox-Stoffwechsel). Medikamente, die die Levels an oxidativem Stress im Parasiten gezielt erhöhen, können diese ROI-Abwehrmechanismen überwältigen und zum Tod des Parasiten führen. Levels an oxidativem Stress können durch Medikamente, die direkte Oxidanzien sind, sowie durch Medikamente, die an Oxidations-Reduktions-Zyklen teilnehmen, erhöht werden, manchmal als sinnloser Redox-Zyklus bezeichnet.
Mögliche Redox-Agenzien
Medikamente
Erreger
Primaquin, Artemisinin-Derivate
Plasmodium
Metronidazol, Tinidazol
Giardia, Entamoeba, Trichomonas
Benznidazol, Nifurtimox
Trypanosoma cruzi
Viele der an Redox-Reaktionen beteiligten Medikamente müssen aktiviert werden, bevor sie gegen ihre Ziel(e) wirksam sind. Zum Beispiel sind Metronidazol und andere Nitroimidazole breit wirkende Medikamente, die eine Vielzahl von anaeroben Bakterien und Protozoen beeinflussen. Diese Medikamente werden durch eine Reduktion der Nitrogruppe zu einem Anionenradikal aktiviert. Das Anionenradikal ist hoch reaktiv und bildet Adjunkte mit Proteinen und DNA, was zu einem Funktionsverlust führt. Insbesondere die Reaktionen mit DNA führen zu Strangbrüchen und Hemmung der Replikation und werden zum Zelltod führen. Die Reduktion von Nitroimidazolen erfordert starke Reduktionsbedingungen, und anaerobe Organismen haben mehr Reduktionspotenzial als aerobe Organismen. Das erklärt die Selektivität dieser Verbindungen für anaerobe Organismen. Mit anderen Worten werden die Medikamente bevorzugt von den Pathogenen aktiviert.
Im Fall von Metronidazol scheint reduziertes Ferredoxin der Hauptelektronendonator für seine Reduktion zu sein (Abbildung). Es besteht eine gute Korrelation zwischen dem Vorhandensein der Pyruvat-Ferredoxin-Oxidoreduktase (PFOR) und der Empfindlichkeit gegenüber Metronidazol. Alle drei von Metronidazol betroffenen Protozoen (Tabelle) haben keine Mitochondrien und verfügen über eine PFOR, die der in vielen anaeroben Bakterien gefundenen ähnelt. Aerobe Organismen mit Mitochondrien verwenden für die Produktion von Acetyl-Coenzym A anstelle der PFOR den Pyruvatdehydrogenasekomplex.
Nitroimidazole (z. B. Benznidazol) und verwandte Nitrofuran-Verbindungen (z. B. Nifurtimox) sind auch wirksam gegen Trypanosoma cruzi. Die für die anfängliche Reduktion dieser Medikamente verantwortlichen Elektronendonatoren sind nicht bekannt, und die Grundlage der Spezifität für den Parasiten ist nicht klar. Beide dieser Medikamente sind etwas toxisch und zeigen keine guten therapeutischen Indizes. Es wird angenommen, dass der Wirkmechanismus von Nifurtimox sinnlose Redox-Zyklen nach seiner Reduktion umfasst, während vermutet wird, dass Benznidazol spezifische Reduktasen hemmt und dadurch die Fähigkeit des Parasiten zur Entfernung von ROI verringert.
Oxidativer Stress und G6PD-Mangel
Verschiedene genetische Krankheiten des Menschen sind dafür bekannt, einen gewissen Schutz vor Malaria zu bieten (siehe angeborene Resistenz). Personen mit Glucose-6-phosphat-Dehydrogenase (G6PD)-Mangel haben niedrigere Spiegel von reduziertem NADPH in ihren Erythrozyten, die für die Aufrechterhaltung von reduziertem Glutathion benötigt werden. Die geringeren Mengen an reduziertem Glutathion führen zu einer erhöhten Empfindlichkeit gegenüber oxidativem Stress, da die Glutathionperoxidase an der Entgiftung von ROI beteiligt ist. Die erhöhten ROI-Spiegel aufgrund des Stoffwechsels des Parasiten in Kombination mit der verringerten Fähigkeit von G6PD-defizienten Erythrozyten, ROI zu entfernen, führen zu einer vorzeitigen Lyse der infizierten Erythrozyten und bieten daher einen gewissen Schutz vor Malaria. Der Parasit muss sich nicht nur vor ROI schützen, sondern auch sicherstellen, dass die Wirtserthrozyten nicht beschädigt werden, bevor der Parasit die erythrozytäre Schizogonie abschließt. Tatsächlich wurde vorgeschlagen, dass der Parasit der Wirtserthrozyte Glutathion zuführen könnte, um deren reduzierende Kapazität zu erhöhen. In ähnlichem Zusammenhang ist die Behandlung mit Primaquin bei G6PD-defizienten Patienten kontraindiziert, da sie hämolytische Anämie verursachen kann. Dies ist wahrscheinlich auf die Fähigkeit von Primaquin zurückzuführen, ROI zu erzeugen, und auf das verringerte Reduktionspotenzial von G6PD-defizienten Erythrozyten.
Arzneimittelresistenz
Mechanismen der ResistenzMutationen im Gen des ZielproteinsErhöhte Produktion des ZielproteinsVerringerung der Medikamentenakkumulation (einschließlich Zunahme des Ausflusses)Inaktivierung des Medikaments
Das Auftreten von Arzneimittelresistenzen begrenzt erheblich das Arsenal an verfügbaren Medikamenten gegen protozoische Erreger. Parasiten haben zahlreiche Möglichkeiten entwickelt, um die Toxizität von Medikamenten zu überwinden (siehe Kasten). Oftmals beinhaltet Arzneimittelresistenz Mutationen im Arzneimittelziel, sodass das Medikament nicht mehr an das Ziel bindet oder es hemmt. Arzneimittelresistenz kann sich schnell entwickeln, wenn eine einzelne Punktmutation Resistenz verleihen kann. Ein weiterer Mechanismus der Arzneimittelresistenz beinhaltet die verstärkte Expression des Zielproteins. Dies kann entweder durch erhöhte Transkription und Translation oder durch Genamplifikation erreicht werden. Dies führt zu einem Bedarf an höheren Medikamentenmengen, um das gleiche Hemmnisniveau zu erreichen. Eine Verringerung der Medikamentenakkumulation oder die Metabolisierung des Medikaments zu nicht toxischen Produkten führen dazu, dass weniger Medikament das Ziel erreicht, und können ebenfalls zur Arzneimittelresistenz beitragen. Arzneimittelresistenz kann auch die Akkumulation von Mutationen in den gleichen oder unterschiedlichen Zielproteinen umfassen, die additive oder synergistische Effekte haben können. Parasiten mit Mutationen oder genetischen Polymorphismen, die eine Verringerung der Empfindlichkeit gegenüber dem Medikament bewirken, werden unter dem Druck des Medikaments selektiert.
Proteine und Mutationen, die mit Arzneimittelresistenz in Verbindung stehen
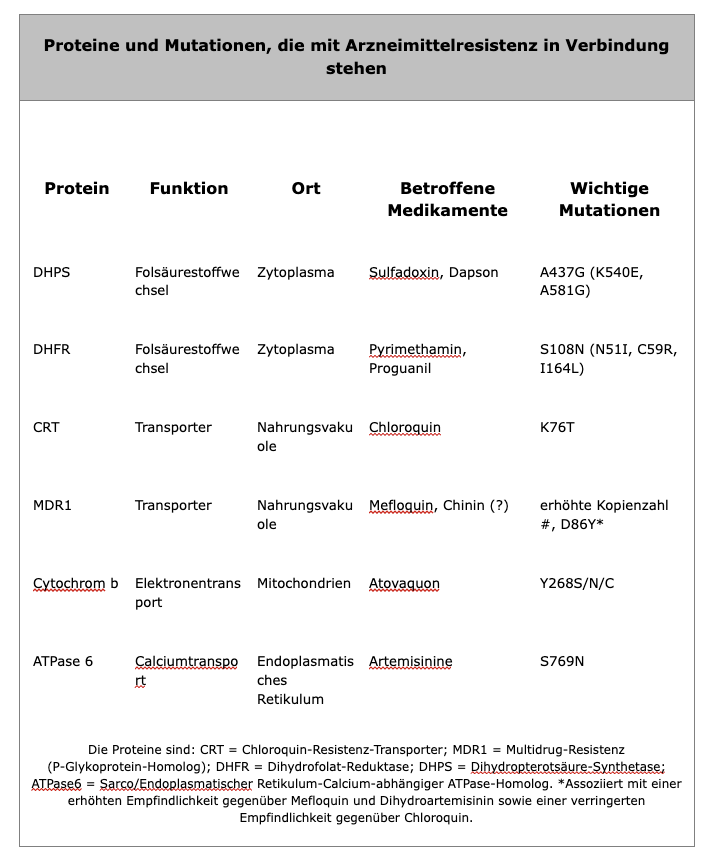
In einigen Fällen wurden spezifische Mutationen mit Arzneimittelresistenz in Verbindung gebracht (siehe Tabelle). Fansidar (SP)-Resistenz korreliert mit spezifischen Mutationen in den von Sulfadoxoin und Pyrimethamin angezielten Enzymen (Dihydropterotsäure-Synthetase und Dihydrofolat-Reduktase). Chloroquinresistenz (siehe unten für weitere Details) wurde mit Mutationen in einem Transporter in der Membran der Nahrungsvakuole (Chloroquin-Resistenz-Transporter, CRT) in Verbindung gebracht. Ein anderer Transporter in der Nahrungsvakuole, das Multidrug-Resistenzgen 1 (MDR1), wurde angedeutet, eine unterstützende Rolle bei der Resistenz zu spielen. Die Grundlage für die Resistenz gegen Mefloquin und Chinin ist nicht klar, aber auch das mdr1-Gen wurde in Zusammenhang gebracht.
Chloroquin. Die Chloroquinresistenz ist mit einer Abnahme der Menge an Chloroquin verbunden, die in der Nahrungsvakuole akkumuliert, dem Wirkort von Chloroquin (siehe oben). Der Mechanismus für diese verringerte Akkumulation ist umstritten. Einige Studien haben gezeigt, dass die Abnahme der Medikamentenakkumulation auf eine Zunahme des Medikamentenabflusses zurückzuführen ist, während andere Studien darauf hinweisen, dass verringerte Mengen an Chloroquin-Akkumulation wichtiger sind. Die Beobachtung, dass Verapamil und verwandte Medikamente das chloroquinresistente Phänotyp umkehren können, hat zu Spekulationen geführt, dass ein ATP-abhängiger Transporter eine Rolle im Medikamentenabfluss und der Chloroquinresistenz spielt, ähnlich der Multidrug-Resistenz (MDR) bei Krebs. Ein MDR-ähnlicher Transporter, der als PfMDR1 bezeichnet wird, wurde auf der Membran der Nahrungsvakuole identifiziert. Es konnten jedoch keine definitive Korrelationen zwischen PfMDR1 und Chloroquinresistenz nachgewiesen werden. Eine unterstützende Rolle von PfMDR1 bei der Chloroquinresistenz kann jedoch nicht ausgeschlossen werden.
Genetische Kreuzungen und Kartierungsstudien zwischen einem chloroquinresistenten Klon und einem chloroquinsensiblen Klon führten zur Identifizierung einer 36 kb-Region auf Chromosom 7, die mit der Chloroquinresistenz in Verbindung steht. Eines der 10 Gene in dieser 36 kb-Region codiert für ein Protein mit 10 transmembranösen Domänen und ähnelt einem Transporterprotein, das Chloridkanälen ähnelt. Das Gen wurde als pfcrt bezeichnet und das Protein ist auf der Membran der Nahrungsvakuole lokalisiert. Mehrere Mutationen im pfcrt-Gen zeigen Korrelationen mit dem Chloroquinresistenz-Phänotyp, und eine Mutation, ein Austausch von Threonin (T) gegen Lysin (K) an Position 76 (K76T), zeigt eine perfekte Korrelation mit der Chloroquinresistenz. Diese Mutationen beeinflussen wahrscheinlich die Akkumulation von Chloroquin in der Nahrungsvakuole, aber der genaue Mechanismus der Chloroquinresistenz ist nicht bekannt. Darüber hinaus hat die Beobachtung, dass die Chloroquinresistenz vergleichsweise wenige Male aufgetreten ist und sich dann verbreitet hat, zu Spekulationen geführt, dass mehrere Gene an der Entwicklung der Resistenz beteiligt sind (siehe weitere Diskussion).
Zusätzliche Literatur
- Foley M and Tilley L (1998) Quinoline-Antimalariamittel. Wirkmechanismen und Resistenz sowie Aussichten auf neue Wirkstoffe. Pharmacology & Therapeutics 79:55
- Hyde JE (2007) Arzneimittelresistente Malaria - ein Einblick. FEBS Journal 274, 4688-4698.
- Ouellette M (2001) Biochemische und molekulare Mechanismen der Arzneimittelresistenz bei Parasiten. Trop Med Int Health 6:874.
- Rosenthal PJ und Goldsmith RS (2001) Antiprotozoale Medikamente. In Grundlagen und klinische Pharmakologie, 8. Auflage. McGraw-Hill Companies Inc. (Online-Ausgabe über Stat!Ref Books in der Tulane Medical Library verfügbar)
- Samuelson J (1999) Warum Metronidazol gegen Bakterien und Parasiten wirksam ist. Antimicrob Agents Chemother. 43:1533.
- Wellems TE und Plowe CV (2001) Chloroquinresistente Malaria. J Inf Dis 184:770.
Original Source is here.
Frequently asked questions

.webp)
New posts to your inbox!
Your submission has been received!